If you want to understand the background to this post, I suggest you read posts 22.12, 22.14, 22.15, 22.20, 22.21, 22.23, 22.24, 23.1 and 23.3 (route 1).
But, for a less mathematical introduction, you can read posts 18.10, 18.16, 22.21 (but not the last paragraph) and 23.2 (route 2).
Most of the statements that appear in this post are justified whichever route you follow.
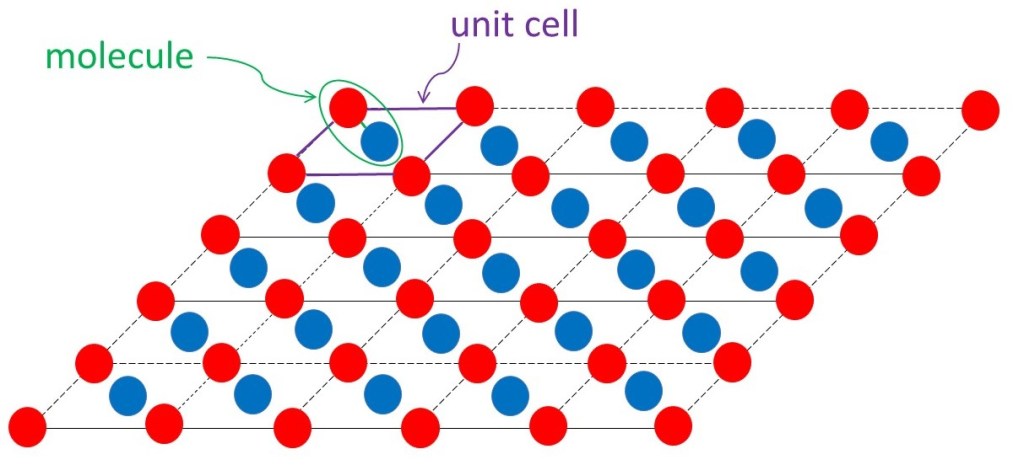
Principles
When a ray (of x-rays), with a single wavelength, λ, passes through a crystal it emerges as many rays. Each ray direction is characterised by three integers (whole numbers) h, k and l. These emerging x-rays are sometimes called Bragg reflections (see post 23.2), often abbreviated to reflections, and the integers are called their Miller indices. The amplitude and phase of a Bragg reflection can be represented by the structure factor, F(h, k, l), for the crystal where
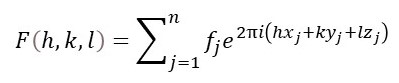
Here fjis called the atomic scattering factor and it represents the x-rays scattered by the j-th atom in a unit cell of the crystal whose position in the unit cell is given by the fractional unit cell coordinates xj, yj and zj (see post 22.21). Atomic scattering factors are known, for each element, because they can be calculated from theory (see post 22.15). The summation, in the equation above, is over all the atoms in a unit cell. The letter i, in the equation above is the square root of -1, so F(h, k, l) is a complex number; this is explained in post 18.16. It is because F(h, k, l) is a complex number that it can represent an amplitude and a phase; this is explained in post 18.17. The letter e has an approximate value of 2.718; further details are given in post 18.15.
In principle, we can record F(h, k, l) experimentally to calculate the distribution of electrons (usually called the electron density), ρ(x, y, z), in a unit cell using the equation below.
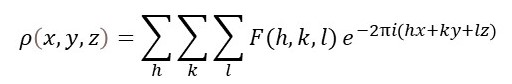
The summation is over all the h, k and l values for the observed reflections. Here x, y and z are the fractional unit cell coordinates at a point in the unit cell (see post 22.21). Since the electrons will be concentrated around atoms, ρ(x, y, z) shows us the positions of atoms in the unit cell. If atoms are close to each other, they are bonded to form molecules;then the positions of the atoms define the molecular structure.
Problems
But there is a fundamental problem. When we use a detector to measure F(h, k, l), we are measuring the energy transmitted by the wave that it represents. This energy depends only on the modulus of F(h, k, l) and not on its phase; this is explained in post 19.8. But to calculate ρ(x, y, z), we need to use the complex number F(h, k, l), that is we need both amplitude and phase information (see post 18.17). This problem is called the phase problem in x-ray crystallography. Fortunately, there are both experimental and statistical methods for solving this problem that I hope to write about in later posts.
The probability that F(h, k, l) will be recorded, for a given incident x-ray direction depends on the scattering angle, ϕ = 2θ, (θ, often called the Bragg angle, was introduced on post 23.2) and how the crystal and detector are moved to record the diffraction pattern. In route 1, this is explained by the probability that a reciprocal lattice point passes through the Ewald sphere. In route 2, this is explained by the probability that a plane of atoms will be in a reflecting position. When the modulus of F(h, k, l) is obtained experimentally, it must be corrected for this effect before we calculate ρ(x, y, z). The correction factor is called the Lorentz factor.
A further correction factor is called the polarisation factor because the modulus of F(h, k, l) obtained experimentally depends on ϕ in a way which depends on the state of polarisation of the incident x-rays. I hope to write about this effect in a future post.
Refinement
When we have calculated ρ(x, y, z), we can identify the positions of the atoms in the unit cell and so calculate the moduli of F(h, k, l) from the first equation in this post.
The final stage of determining the positions of the atoms in a unit cell and, therefore, in a molecule, is the process of refinement. In refinement the positions of the atoms are systematically varied to obtain the best fit between the observed and calculated values of the moduli of F(h, k, l). Mathematically, this is analogous to curve-fitting, explained in the appendix to post 19.2.
During refinement, we can also allow for the effect that thermal vibrations have on the positions of atoms in a crystal (see post 16.38). Vibration of an atom smears out its electron density distribution, so the calculated values of fj are not exact. We can allow for this problem by modifying fj to
f’j = fjexp(-K2σj2/2).
Here σj is the standard deviation calculated over all the positions for the j-th vibrating atom and exp(x) is a different way of writing ex; I’ve used it here to make the equation easier to read. Also
K = (4π/λ)sin(ϕ/2) = (4π/λ)sinθ.
So, now our expression for the structure factor becomes
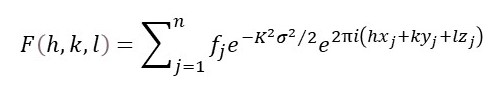
And refinement consists of varying the positions and standard deviations for each atom in the unit cell. But most text books on x-ray crystallography write the above equation as
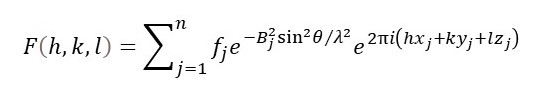
where Bj is called the isotropic temperature factor, for the j-th atom; it is isotropic because it assumes that the standard deviations are the same for all vibration directions. If we don’t make this assumption, there are 6 anisotropic temperature factors for each atom to be included in the refinement.
There are many statements in the paragraph above that I have not justified but I hope to write about them in a future post.
At the end of refinement, we know the positions of the atoms in a molecule (when it is in a crystal), and how much they are affected by thermal vibrations. Results from many structures determinations tell us the values of bond lengths and bond angles for covalent bonds between different elements.
Follow-up posts
23.10 Frequency analysis and x-ray crystallography
24.2 Thermal vibrations
