Before you read this, I suggest you read post 21.4; it would also be useful to read post 21.6.

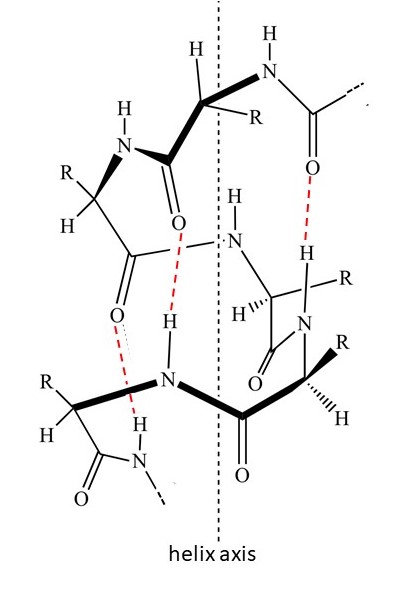
Some protein molecules can form a three-dimensional structure call the α-helix (α is the Greek letter “alpha”), shown in the picture above. In this picture, the coloured balls represent different atoms: black for carbon, white for oxygen, red for oxygen and blue for nitrogen. The α-helix shape has 3.6 amino acid residues in each turn of the helix. It is stabilised by hydrogen bonds between different residues, as shown (by red dashed lines) in the picture below. The picture tries to make the structure look three-dimensional by having dark bonds at the front and tapering some bonds; dashed bonds tht are tapered point behind the plane of the picture.
Some proteins form fibres in which the molecules are aligned – in the same way that stretched rubber molecules are aligned. Hair is an example of this type of fibrous protein that contains molecules of the protein keratin. The English biophysicist William T Astbury (1898-1961) recorded x-ray diffraction patterns from many fibrous proteins, in the same way that x-ray diffraction patterns can be recorded from stretched rubber. He obtained three types of pattern; one was from collagen – the other two he called α and β (β is the Greek letter “beta”) . The α pattern showed that the three-dimensional structure of the molecules had a spatial frequency, in the direction of the fibre, corresponding to a repeat distances of 0.15 nm, as explained in appendix 1. This observation was explained by the American chemist Linus Pauling (1901-1994).
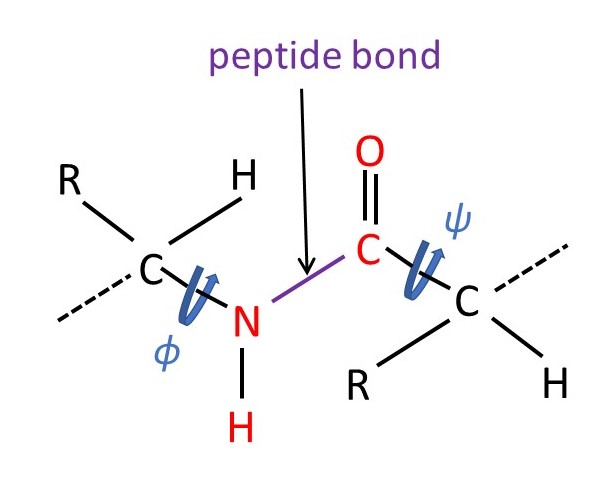
Pauling realised that the red atoms, in the picture above, must all be in the same plane, because the peptide bond (defined in the picture above) is a bit like a double bond for the reasons described in appendix 2. Rotation about a double bond bond is not allowed, so the red atoms are fixed in a plane. The three-dimensional shape of any protein chain can then be described by the angles ϕ and ψ shown in the picture above. He also realised that protein molecules that can be aligned must have a regular shape, meaning that ϕ and ψ must have the same values all along the protein chain. The next step was to build scale molecular models of a protein chain. This model building depends on two observations: (1) bonds between the same atoms in different molecules have the same length and (2) the angle between the same bonds is the same in different molecules. For example, the C=O double bond has a length of 0.120 nm and the N-C bond length is 0.146 nm. The bond angle between C=O and C-N is 125o. These results can be obtained by determining the positions of atoms in molecules using the technique of x-ray crystallography.
Pauling found that he could make his models into a non-integral helix with a pitch of 0.51 nm and 3.6 peptide resides per 360o turn: that is with an axial rise-per-residue of 0.51/3.6 = 0.15 nm. So, he was able to explain Astbury’s α x-ray diffraction pattern and called this helix the α-helix. He used the same method to also explain the β pattern. However, he couldn’t tell whether the α-helix was right or left-handed. When x-ray crystallography was used to find the three-dimensional structures of globular proteins, many were found to have α-helical regions. And they were all right-handed. So we now believe that the α-helix is right-handed.
This model-building approach was later used to determine the three-dimensional structures of nucleic acids and polysaccharides in the x-ray diffraction patterns from gels in which the polymer chains were oriented by shear. I have simplified the history of this technique, sometimes called x-ray fibre diffraction, in this post. But I provide a few more details in appendix 3.
Related posts
Follow-up posts
25.2 Sigma bonds and pi bonds (furher explanation of restricted rotation about peptide bon)
22.14 X-ray diffraction
Appendix 1: x-ray diffraction by oriented polymers
It might be useful to read post 19.20 before reading this appendix.
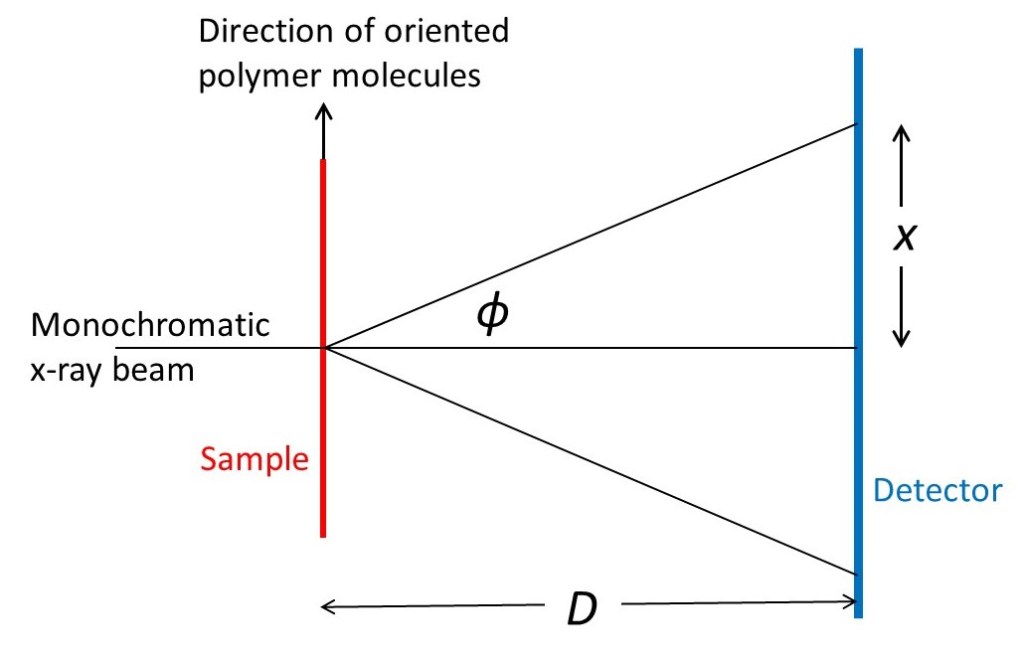
The picture above shows a narrow beam of x-rays with a single wavelength (monochromatic x-rays) perpendicular to a sample (like a fibre) containing oriented polymer molecules; the molecules are oriented parallel to the axis of the fibre. A plane detector (like a sheet of photographic film) is perpendicular to the direction of the beam, a distance D from the specimen. X-rays scattered through an angle φ meet the detector a distance x from the position of the undeflected beam. From the definition of the tangent of an angle
tanφ = x/D ⇒ φ = arctan(x/D) (1)

Now suppose we have an array of scattering objects, like the monomers in a polymer, an axial distance d apart, as shown in the picture above. We are first going to think about scattering in the φ direction by the atoms at A and B; AC is drawn perpendicular to the wave scattered by B. Since the direction of propagation of the scattered wave makes an angle φ with the horizontal (in the picture), AC makes an angle φ with the vertical direction between the scatterers. The condition for constructive interference is that the length of BC is a whole number of wavelengths, so that (from the definition of the sine of an angle)
dsinφ = nλ ⇒ d = nλ/sinφ (2)
where n is an integer (whole number) and λ is the wavelength of the x-rays.
We can measure x and D in an experiment and use equation 1 to calculate φ. If we know λ (usually 0.154 nm because it is easy to produce monochromatic x-rays with this wavelength) we can use equation 2 to calculate d, where n = 1 for the first point of non-zero scattered intensity above the undeflected beam. In Astbury’s α pattern, d = 0.15 nm.
Appendix 2: why is the peptide bond a bit like a double bond?
Pauling explained this result using the valence bond theory. But I think it’s easier to use the molecular orbital theory.
The second bond in C=O is formed by overlap of p orbitals perpendicular to the plane of the third picture in this post. The lone pair of electrons on the N atom are in a p orbital that is also perpendicular to this plane. So all three p orbitals (on the O, C and N atoms) can overlap to form a delocalised molecular orbital. This delocalised molecular orbital would be destroyed by rotation about the peptide bond, because the p orbitals would no longer overlap..
Appendix 3: a brief history of determining three-dimensional molecular structures by x-ray fibre diffraction
Astbury realised that the protein chain would need to be helical, to explain his α x-ray diffraction pattern, and that the β pattern could be explained by uncoiling of the helix, in the 1930s. He also believed that the helix could be stabilised by hydrogen bonds between the C=O and N-H groups of different amino acid residues. So, it appears that he anticipated many of Pauling’s results by about 20 years. The American chemist Maurice L Huggins (1987-1981) proposed a structure very similar to the α -helix about 10 years before Pauling produced his model.
The British chemist Charles W Bunn (1905-1990) used methods that were similar to those of Pauling to determine the three-dimensional structure of the polymer chain in stretched rubber and simpler polymers in the 1940s.
Pauling developed his ideas in collaboration with the American chemist Robert B Corey (1897-1971) and the American physicist Herman R Branson (1914-1995). It is possible that Branson’s contribution to this work was much greater than is generally supposed.
Subsequently x-ray fibre diffraction was used to investigate nucleic acids, polysaccharides and a variety of synthetic polymers. Much of the early work didn’t involve calculating the intensity distribution of the diffraction pattern as a direct test of the proposed molecular models. This approach was developed by the British biophysicist Maurice Wilkins (1916-2004) and his colleagues in their work on DNA. They were then able to adjust the model to get the best fit between the observed and calculated intensity distributions. In this way they wer able to refine the original model. The British scientist Struther Arnott (1934-2013) and his colleagues at Kings College London and, later, Purdue University developed a computer modelling technique to refine molecular models in this way, while constraining them to have the measured pitch and axial rise-per-residue. This technique has been applied to rubber, proteins, nucleic acids, polysaccharides and several synthetic polymers. A modification of this constrained modelling technique can also be used to build the initial models. The advantage of computer modelling is that the coordinates of the atoms in the molecule can be defined with much greater precision than would be possible with a physical model.
